
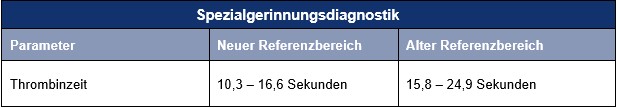
Gerinnungsdiagnostik: Thrombinzeit – Änderung des Referenzbereichs
Ab 1. Oktober 2025 werden wir für die Thrombinzeitbestimmung (entsprechend den Empfehlungen der Herstellerfirma WERFEN) eine andere Reagenz-Anlösung verwenden. Hierdurch ändert sich lediglich der Referenzbereich für den Parameter Thrombinzeit. Dieser wird für Sie wie gewohnt mit auf dem Befund ausgewiesen.
Bei Rückfragen kontaktieren Sie bitte die laborärztlichen Kolleginnen und Kollegen unter Tel. 030.820 93-449.
Auswertung der Resistenzdaten von Urinuntersuchungen aus dem Jahr 2024
Die Kenntnis der lokalen Resistenzsituation ist für die Wahl einer effektiven empirischen Antibiotikatherapie von entscheidender Bedeutung. Grundlage der vorliegenden Resistenzstatistik sind Urinuntersuchungen von ambulanten Patientinnen und Patienten, getrennt nach Erwachsenen (Tabelle 2) und Kindern (Tabelle 3). Die „I“-Kategorie (= sensibel bei erhöhter Exposition) wird in dieser Resistenzstatistik zu den empfindlichen Isolaten gezählt, da bei hoher Dosierung oder durch Anreicherung des Antibiotikums am Infektionsort, etwa durch eine hohe Konzentration im Urin, mit einer Wirksamkeit der Substanz gerechnet werden kann. Da es seitens des Europäischen Komitees zur Antimikrobiellen Empfindlichkeitstestung (EUCAST) keine standardisierten Vorgaben zur Bestimmung der Empfindlichkeit für alle Erreger-Antibiotika-Kombinationen gibt, kann nicht für alle Substanzen eine Resistenzbestimmung erfolgen. Dies liegt an noch fehlenden klinischen Daten zur Wirksamkeit bestimmter Substanzen oder an einer natürlichen (intrinsischen) Resistenz einiger Erreger.
Gemäß der AWMF-S3-Leitlinie¹ gilt ein Antibiotikum für die empirische Therapie einer unkomplizierten Zystitis bei Empfindlichkeitsraten unter 80 % als ungeeignet. Für die empirische Behandlung einer akuten unkomplizierten Pyelonephritis sollte die lokale Empfindlichkeitsrate sogar mindestens 90 % betragen.¹
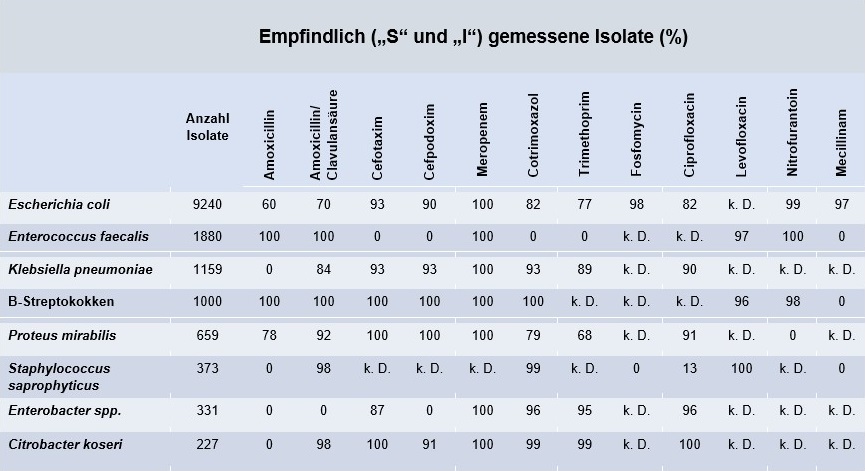
Am häufigsten nachgewiesen wurde Escherichia coli (E. coli) mit einer hohen Empfindlichkeitsrate gegenüber Fosfomycin, Nitrofurantoin und Mecillinam. Bei Trimethoprim ist jedoch der Anteil resistenter E. coli-Isolate inzwischen so hoch, dass der Einsatz ohne vorherige Resistenztestung nicht empfohlen werden kann. Die Resistenzsituation für Cotrimoxazol ist mit einer Empfindlichkeitsrate von 82% bei Erwachsenen und 81 % bei Kindern gerade noch akzeptabel für einen empirischen Einsatz. Dieser Anteil steigt jedoch bei rezidivierenden Harnwegsinfektionen an und im deutschlandweiten Vergleich zeigt sich ein zunehmender Trend der Resistenz.1,2 Daher wird in der aktuellen Leitlinie Cotrimoxazol nicht mehr als Medikament der ersten Wahl für die empirische Therapie einer unkomplizierten Zystitis empfohlen.¹
Seit 2021 ist die Rate an 3MRGN E.coli-Isolaten(d.h. multiresistente gram-negative Bakterien mit einer Resistenz gegen Piperacillin, Cephalosporine der 3. Generation und Chinolone) deutlich angestiegen: von 3,0 % auf aktuell 5,6 % bei Erwachsenen und von 3,3 % auf 6,8 % bei Kindern. 4MRGN-Isolate (d.h. mit zusätzlicher Resistenz gegen Carbapeneme) sind bislang nur vereinzelt im ambulanten Bereich nachweisbar.
Diese Daten verdeutlichen die Notwendigkeit eines rationalen Antibiotika-Einsatzes im ambulanten Bereich zur Eindämmung weiterer Resistenzentwicklungen.
---
Tabelle 2: Empfindlichkeit der häufigsten Bakterien in Urinproben bei Erwachsenen (Ambulante Patientenversorgung, 2024)
k. D.= keine Daten
---
Tabelle 3: Empfindlichkeit der häufigsten Bakterien in Urinproben bei Kindern (Ambulante Patientenversorgung, 2024)
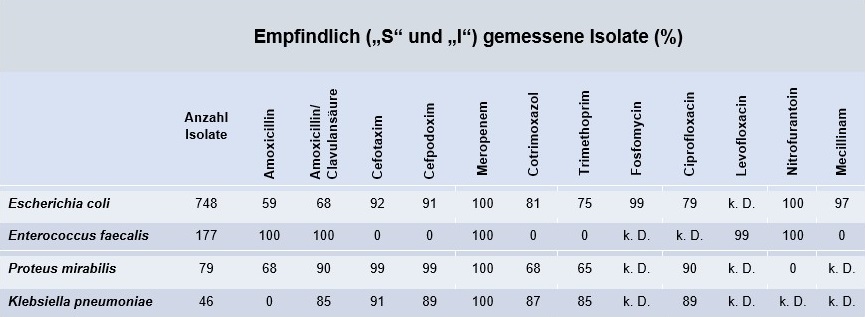
Hinweis: Eine Resistenzstatistik konnte aufgrund der geringen Anzahl an Nachweisen nicht für alle Bakterien erstellt werden.
k. D.= keine Daten
---
---
Literatur:
1. AWMF S3 Leitlinie: Epidemiologie, Diagnostik, Therapie, Prävention und Management unkomplizierter, bakterieller, ambulant erworbener Harnwegsinfektionen (HWI) bei Erwachsenen. Langversion 3.0-Stand April 2024. AWMF Registernummer: 043-044
2. Robert Koch-Institut: ARS (Antibiotika-Resistenz-Surveillance), https://ars.rki.de, Datenstand:12.06.2025
eGFR-korrigiertes NT-proBNP
Das N-terminale pro Brain natriuretische Peptid (NT-proBNP) gehört heute zur Standarddiagnostik bei akuter und chronischer Herzinsuffizienz – sowohl initial als auch im Rahmen der Verlaufskontrolle. Es ist jedoch bekannt, dass erhöhte NT-proBNP- Konzentrationen im Serum nicht ausschließlich auf kardiovaskuläre Ursachen zurückzuführen sind. Besonders eine eingeschränkte Nierenfunktion kann zu erhöhten Werten führen, da
NT- proBNP überwiegend renal eliminiert wird.
Dieser inverse Zusammenhang zwischen glomerulärer Filtrationsrate (eGFR) und NT-proBNP- Konzentration birgt das Risiko einer Fehlinterpretation der kardialen Funktion bei Patientinnen und Patienten mit akuten oder chronischen Nierenerkrankungen.
Um dem entgegenzuwirken, besteht die Möglichkeit, NT-proBNP-Werte anhand der von Luchner et al.¹ vorgeschlagenen Formel zu korrigieren:
NT-proBNP adjusted = NT-proBNP/e1.892-0.025 x eGFR
Dabei ist zu beachten, dass diese Korrektur nur bei einer berechneten eGFR < 75 ml/min Anwendung finden sollte. In der zugrunde liegenden Studie wurden zudem weder Dialysepatienten noch Patienten mit einer eGFR
< 15 ml/min berücksichtigt. Darüber hinaus ist die Formel aus der Untersuchung einer männlichen Population abgeleitet worden; vergleichbare Daten für weibliche Patientinnen sind bislang nicht publiziert
Beispiel:
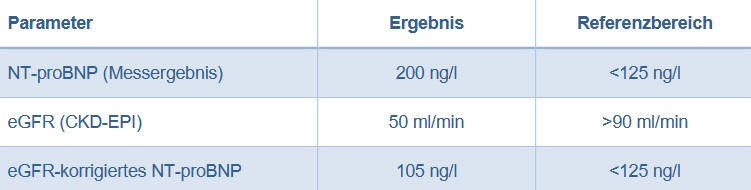
Ab dem 01.06.2025 können Sie das eGFR-korrigierte NT-proBNP über star.net® anfordern. Auf dem Befund werden sowohl der gemessene als auch der rechnerisch korrigierte NT- proBNP-Wert erscheinen.
---
Literatur:
1. Luchner A et al. Improvement of the cardiac marker N-terminal-pro brain natriuretic peptide through adjustment for renal
function: a stratified multicenter trial. Clin Chem Lab Med 2010; 48(1):121–128.
Apolipoprotein E und Lecanemab (Leqembi®)
Der monoklonale Antikörper Lecanemab ist gegen die, für die Entwicklung einer Alzheimer-Krankheit (AD) pathophysiologisch wichtigen, Amyloid-Plaques gerichtet. Das Medikament kann den Krankheitsverlauf im frühen Stadium der AD verlangsamen.
Unerwünschte Nebenwirkungen können hier u. a. aus einer übersteigerten Immunreaktion resultieren. Diese lokalen Reaktionen im Hirngewebe werden unter dem Oberbegriff „amyloid-related imaging abnormalities“ (ARIA) zusammengefasst. Sie präsentieren sich z. B. als Ödem (ARIA-E) oder Mikroblutungen (ARIA-H) im Hirnparenchym. Das Risiko, ARIA im Rahmen einer Lecanemab-Therapie zu entwickeln, steigt mit zunehmender Plaquedichte und ist mit dem APOE-Genotyp assoziiert.
Im November 2024 hat der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Arzneimittel-Agentur (EMA) eine positive Empfehlung für die Zulassung von Leqembi® mit dem Wirkstoff Lecanemab für die Alzheimer-Therapie ausgesprochen. Die entsprechende Zulassung erteilte die Europäische Kommission am 15. April 2025. Zugelassen ist Lecanemab zur Behandlung einer leichten kognitiven Beeinträchtigung (Gedächtnis- und Denkstörungen) oder einer leichten Demenz in frühen Stadien der AD.
Vor dem Beginn der Behandlung mit Lecanemab ist ein Gentest erforderlich.
Lecanemab ist für diejenigen Alzheimer-Patienten indiziert, die „nur eine oder keine Kopie des APOE4-Allels“ aufweisen. Dies trifft für rund 85 % aller Menschen mit Alzheimer zu. Bei ihnen ist die Wahrscheinlichkeit für ARIA geringer als bei Menschen mit APOE4-Homozygotie (ca. 15 % der Betroffenen).
Eine Testung auf den APOE4-Status sowie eine entsprechende Aufklärung sollte vor Einleitung der Behandlung durchgeführt werden, um das Risiko der Entwicklung einer ARIA abzuschätzen.
https://ec.europa.eu/health/documents/community-register/2025/20250415164782/anx_164782_en.pdf
Im Labor 28 führen wir die molekulargenetische Untersuchung des APOE-Gens (APOE-Genotypisierung) mittels Real-Time PCR durch. Für diese Untersuchung wird EDTA-Blut und eine Einwilligung nach GenDG benötigt. Bei der Anforderung bitten wir um den Hinweis auf die geplante Lecanemab-Therapie.
Hämatologie: BSG (Blutsenkungsgeschwindigkeit)-Bestimmung – neue Gerätegeneration ALIFAX Test1 2.0
 Ab 17.03.2025 wird für die automatisierte BSG-Bestimmung (Methode: quantitative Kapillarphotometrie) eine neue und aktualisierte Gerätegeneration in unserem Labor eingesetzt. Der Hersteller (Fa. Alifax S.p.A.) gibt an, dass die Aggregation der Erythrozyten dabei stark mit den Endpunkt-Ergebnissen der klassischen Westergren-Methode korreliert, jedoch nicht von Interferenzen betroffen ist.
Ab 17.03.2025 wird für die automatisierte BSG-Bestimmung (Methode: quantitative Kapillarphotometrie) eine neue und aktualisierte Gerätegeneration in unserem Labor eingesetzt. Der Hersteller (Fa. Alifax S.p.A.) gibt an, dass die Aggregation der Erythrozyten dabei stark mit den Endpunkt-Ergebnissen der klassischen Westergren-Methode korreliert, jedoch nicht von Interferenzen betroffen ist.
Unsere Validation konnte zeigen, dass insbesondere außerhalb des Referenzbereichs (Frauen >= 25 mm/h, Männer >= 20 mm/h) höhere Werte (mittlere Abweichung bis zu 40 %) im Gegensatz zu den Vorgänger-geräten (siehe Abbildung) gemessen werden. Wie vom Hersteller angegeben, korrelieren diese höheren Werte besser mit der klassischen Westergren-Methode.
Diese Veränderung sollte insbesondere bei einer Longitudinalbeurteilung mitberücksichtigt werden.
Gerinnungsdiagnostik: Multiplate® Analyzer
Ab 03.03.2025 benötigen wir für die Multiplate-Untersuchung ein Hirudinröhrchen (bisher Li-Heparin- Röhrchen). Diese können Sie gerne wie gewohnt in unserem Materiallager bestellen.
Die Präanalytik für die Multiplate-Untersuchung verändert sich durch die Umstellung nicht. Eine Blutentnahme im Labor 28 ist für die optimale Bestimmung weiterhin zu präferieren. Sollten Sie in Ihrer Praxis das Blut abnehmen, so ist die vollständige Befüllung des Hirudinröhrchens (1,6 ml) von großer Bedeutung.
Verbesserung der Diagnostik von EHEC (enterohämorrhagische Escherichia coli) im Stuhl
Nachdem wir in den vergangenen Jahren bereits die PCR-Diagnostik von Parasiten und Viren im Stuhl von ELISA-Verfahren auf sensitivere Nachweise mittels PCR umgestellt hatten, geschieht dasselbe nun auch ab 17.02.2025 für den für Nachweis von EHEC (enterohämorrhagische Escherichia coli).
Bei den Anforderungen „Stuhl auf pathogene Keime“ oder „Stuhl auf E+R“ wird automatisch eine PCR-Untersuchung für den EHEC-Nachweis durchgeführt. Bislang wurden diese Proben zunächst mit einem ELISA auf das Vorliegen von Shigatoxin gescreent und hierin positive Proben dann am Folgetag mittels PCR untersucht. Nur bei Kindern < 3 Jahren wurde bereits eine PCR durchgeführt, um neben EHEC auch EPEC zu erkennen, die v. a. in dieser Altersgruppe relevante Gastroenteritiserreger sind.
Nach der Umstellung werden zukünftig alle Proben mit der sensitiveren PCR untersucht, und wir können Ihnen außerdem die Befunde schneller mitteilen, da die bisherige Zweistufigkeit entfällt. Selbstverständlich kann die Untersuchung auf EHEC auch weiterhin einzeln angefordert werden.
Diese Umstellung ist nicht nur eine deutliche Verbesserung der Diagnostik durch die Verwendung einer sensitiveren Nachweismethode als zuvor sowie die schnellere Befundübermittlung, sondern auch wirtschaftlicher, da bei den Anforderungen „Stuhl auf pathogene Keime“ oder „Stuhl auf E+R“ die EHEC-PCR neben der ebenfalls durchgeführten Norovirus-PCR mit 7,23 EUR (GOP 32853, bei Angabe der 32006 gegenüber der KV nicht budgetbelastend) verglichen mit dem zuvor durchgeführte ELISA (GOP 32705: 8,56 EUR) berechnet wird.